Why do I see a high % of Duplicate Sequences (FastQC) in my QuantSeq data?
QuantSeq generates inherently low-complexity libraries; Reverse transcription is initiated by oligodT priming, which anchors the first strand synthesis to the very 3' end of the transcript. Furthermore, the protocol yields libraries with mean insert sizes of 203 – 324 bp. Consequently, the great majority of library fragments corresponding to a particular gene come from a region <400 nt immediately upstream of the poly(A) tail. This reduces the likelihood of any given read to have unique mapping coordinates, even though the second strand synthesis is initiated by random priming.
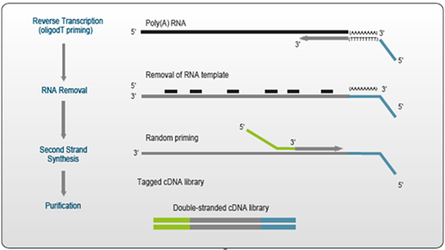
Figure 1. Schematic Overview of QuantSeq library generation.
The plot below shows the distribution of % Duplicate Reads, as reported by FASTQC, from Lexogen's internal runs as reference.
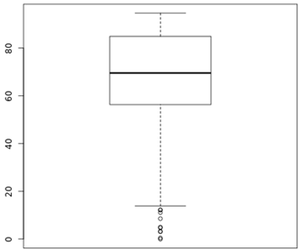
Figure 2. Distribution of % Duplicate reads from Lexogen runs.