Do my QuantSeq FWD-UMI reads look okay?
QuantSeq FWD-UMI libraries contain a 4 nt spacer located between the 6 nt UMI sequence and the start of the read from the random priming sequence that initiates second strand synthesis. The spacer has the same sequence (TATA) in all library fragments, which generates low diversity signals for cycles 7-10 of Read 1. This low diversity signature is visible in both sequencing run results (e.g., SAV files, or basespace run results) and FastQC analysis results.
The figures below show the FastQC per base sequence content for QuantSeq FWD (standard, no UMI, Fig. 1) and QuantSeq FWD-UMI (Figure 2) libraries.
We recommend adding a minimum of 5 – 15 % PhiX (maximum 30 %) spike-in when sequencing QuantSeq FWD-UMI libraries on all Illumina instruments. The higher PhiX percentage corrects for the low diversity of the spacer.
ATTENTION! The optimal PhiX spike-in percentage may differ between instruments. 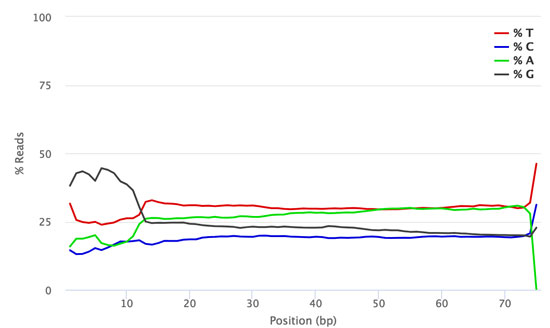
Figure 1 | FastQC Per Base Sequence Content profile for QuantSeq FWD library pool sequenced on NextSeq 500 (no UMIs).
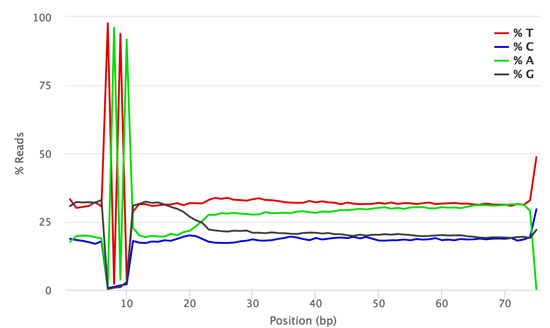
Figure 2 | FastQC Per Base Sequence Content profile for QuantSeq FWD-UMI library pool sequenced on NextSeq 500.
NOTE: The QuantSeq FWD-UMI data analysis pipeline provided by Lexogen trims the UMI and spacer from the beginning of Read 1 before read Quality Control is performed. Therefore, FastQC reports for raw and trimmed reads look the same as for Standard QuantSeq FWD libraries (without UMIs, see https://lexogen-customerportal.atlassian.net/l/c/eWYn0JSt for pipeline details).