Why do I see a peak at ~ 200 bp in my libraries?
What is this peak?
The presence of a peak at ~200 bp in QuantSeq FWD libraries can be an indication of mishybridization.
Mishybridization (i.e., mis-priming of the oligo(dT) primers) can happen as a result of deviation from optimal incubation temperatures during first strand cDNA synthesis (i.e., reverse transcription).
In this case, mis-priming may occur in A-rich regions (e.g., ribosomal RNAs) other than the polyA tail of mRNA molecules. As these non-specific inserts are small in size, these are efficiently amplified, hence, the presence of distinct peaks on the library traces (see Figure below). Other A-rich regions (present in various RNA molecules, including mRNAs) could also be captured and amplified in a similar way.
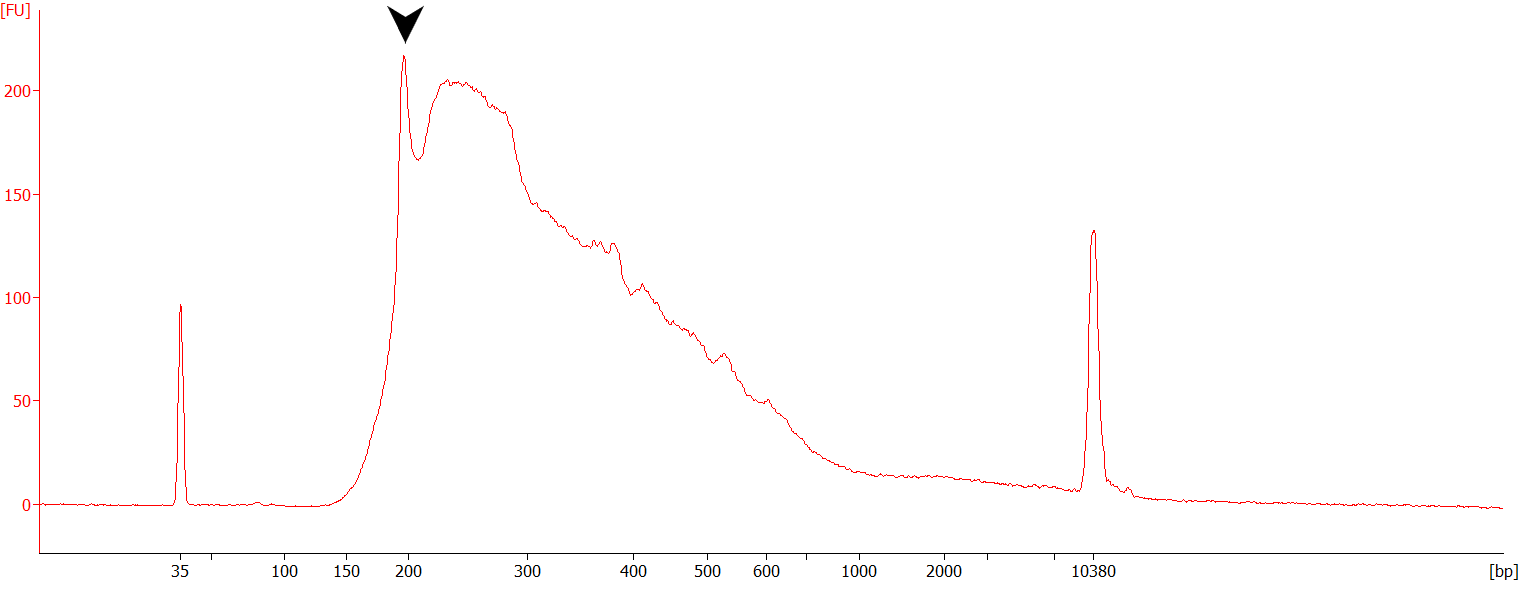
Figure | Example of a QuantSeq FWD library prepared from 50ng of Universal Human Reference RNA (UHRR) and showing sign of mishybridization (peak at ~200 bp corresponding to rRNAs, black arrow).
How do I prevent mishybridization?
To prevent mishybridization, we recommend the following:
Pre-heat the thermocycler block at 42°C.
Keep the RNA / FS1 reaction mix at 42 °C on the thermocycler block after the denaturation step (see steps 2-4 of the User Guide detailed protocol).
Pre-warm the FS2 / E1 mastermix at 42°C before adding to the RNA / FS1 mix (step 3).
Keep the RNA/FS1 mix at 42°C when adding the FS2 / E1 mastermix. Mix well and spin down before continuing to step 4.
NOTE: When using low input/degraded samples, since step 2 is skipped, the FS1 / FS2 / E1 mastermix should be pre-warmed at 42°C before being added to the RNA samples (step 3). We also recommend keeping your RNA samples at RT for a couple of minutes before adding the mastermix to prevent mis priming of the primers before the incubation starts (Step 4).
All centrifugation steps should always be carried out at room temperature (never at 4°C)!
TIP: Raising the reaction temperature to 50°C for reverse transcription could also help prevent mis-priming. However, this can also result in a reduced yield overall and the qPCR assay should be used to ensure the libraries are amplified to provide sufficient yields for sequencing.
What impact does mishybridization have on my results?
Mishybridization products can result in a higher proportion of intronic and ribosomal RNA (rRNA) reads in the sequencing output. As these non-specific inserts are small in size, these are efficiently amplified, resulting in an elevated non-specific read fractions in your final libraries.
Unfortunately, it is not possible to estimate the proportion of mishybridization products present in a library, before sequencing it.
Should I sequence libraries with a mishybridization peak?
Libraries with a mishybridization peak can be sequenced. However, please keep in mind that you may observe unwanted reads (such as ribosomal and intronic sequences) in your sequencing output. The final proportion of these reads will depend on the severity of the library contamination due to the mishybridization.
To ensure maximum data quality, libraries should best be reprepped, taking care of handling temperatures to avoid mishybridization events. If not possible, we recommend increasing the sequencing depth to compensate for the reads taken-up by the rRNAs and/or other sequences (e.g., 5 - 10 M reads per sample).
How to choose between continuing with sequencing vs. reprepping the libraries?
Reasons to re-prep libraries with mishybridization:
Sufficient amounts of RNA samples are available.
The results must be compared with previous libraries, or other library preps that do not show evidence of mishybridization.
Sufficient time / kit reagents / budgets are available to facilitate re-prepping.
Data quality is more important than time / cost.
Additional sequencing depth cannot be accommodated.
Reasons to sequence libraries with mishybridization:
Precious samples that cannot be reprepped.
All samples within the batch / project show similar mishybridization features.
Results are stand-alone and won’t be compared to other sample batches or projects.
Insufficient time / budget / reagents available to reprep libraries.
Additional noise / deviations in data quality can be tolerated.
Additional sequencing depth can be accommodated.
NOTE: The decision of whether or not to sequence libraries with mishybridization signatures resides with the end user. Lexogen provides no guarantees or minimum data quality thresholds for libraries that display mis-hybridization peaks.
If I don’t suspect this peak to be mishybridization, what else could it be?
If this peak is unlikely to be due to mishybridization and the temperatures during First-Strand Synthesis were followed correctly, it may indicate one of the following:
1. The peak is sample-specific.
To assess this, prepare a control library in parallel - ideally using Universal Human Reference RNA. If the ~200 bp peak does not appear in the control library, it is likely that the peak represents an abundant, sample-specific transcript or RNA fragment unique to your species or sample type.
2. The peak is an artifact of TapeStation analysis (e.g., peak fronting).
When libraries are QC’d on the TapeStation, a distorted or unexpected peak can occur due to peak fronting. This happens when DNA fragments of similar but not identical sizes migrate closely and merge, forming a broadened or asymmetric peak. This artifact is more common in libraries prepared from low-input RNA or libraries with overall low concentrations.
To help determine whether peak fronting is occurring, consider the following:
Peak shape: Peak fronting typically produces a skewed peak with a steeper leading edge and a shallower trailing edge rather than a clean, Gaussian shape.
Library concentration: Low-concentration libraries are more prone to these artifacts.
Behavior across replicates:
Artifacts often vary between runs or wells.
Artifacts often appear inconsistently across technical replicates.
True biological peaks are typically consistent across technical replicates prepared from the same RNA.
Because TapeStation artifacts can be instrument- or run-specific, if you suspect peak fronting, it’s helpful to QC the same libraries on an alternative platform (e.g., Bioanalyzer, Fragment Analyzer, or capillary electrophoresis). If the ~200 bp peak persists across platforms, it is more likely to be biological; if it disappears, it was likely a TapeStation artifact.